Afspraken
-
Komt u voor de eerste keer op afspraak? Neem dan eerst contact op met het secretariaat. Zo kunnen we voorafgaande onderzoeken buiten UZ Leuven opvragen of eventueel nog een noodzakelijk onderzoek plannen vóór uw afspraak.
-
+32 16 34 34 91 - werkdagen van 9 tot 12 uur en van 13 tot 14.30 uur
-
Al patiënt bij ons? Vraag uw attest of voorschrift aan.
-
Voor (huis)artsen: stuur uw verwijsbrief via de eHealthBox met als bestemmeling “Bloedings- en vaatziekten – U.Z. Leuven".
Stoornis in stolling
Bloed bevat verschillende eiwitten die belangrijk zijn voor een goede bloedstolling. Ieder eiwit, ook wel stollingsfactor genoemd, heeft een eigen specifieke functie in de vorming van een stevige bloedklonter aan de vaatwand bij een vaatdefect.
Bij hemofilie A ontbreekt het eiwit factor VIII (8) volledig of gedeeltelijk. Bij een bloeding wordt daardoor een bloedklonter gevormd die minder stabiel is.
Er zijn 3 graden van hemofilie A, gebaseerd op de aanwezigheid van factor VIII in het bloed:
| Ernstige hemofilie | FVIII- gehalte kleiner dan 1% van het normale |
| Matig ernstige hemofilie | FVIII gehalte tussen 1 en 5% van het normale |
| Milde hemofilie | FVIII tussen 5 en 50% |
De normale bloedwaarden voor factor VIII situeren zich tussen 50 en 150%.
Diagnose
Hemofilie kan gediagnosticeerd worden via:
- De familiale geschiedenis. De arts zal in een eerste gesprek, naast de specifieke vragen over klinische symptomen en bloedingen bij uzelf, ook vragen stellen naar bloedingen bij ouders, grootouders en andere familieleden.
- Laboratoriumtesten: de stolling wordt gecontroleerd en het factorgehalte in het bloed bepaald.
- Een bloedproef: het erfelijk defect dat verantwoordelijk is voor de hemofilie A wordt opgespoord.
Erfelijkheid
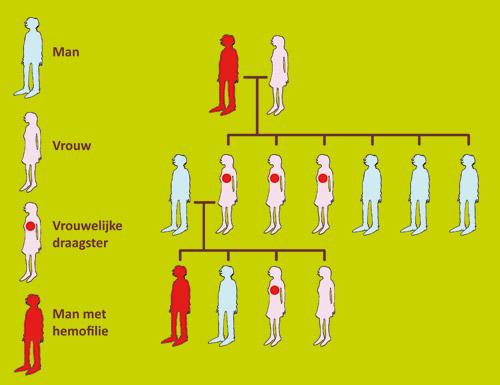
Hemofilie A is overwegend een aangeboren erfelijke aandoening.
Het verantwoordelijke gen bevindt zich op X-geslachtschromosoom.Dat betekent dat het door de moeder, die draagster is van hemofilie, wordt overgedragen.
- Hemofilie A treft enkel jongens (1 op 5.000). Zij hebben namelijk maar één X-chromosoom, dat zij van hun moeder krijgen.
- Zonen van vrouwen die draagster van hemofilie zijn, hebben één kans op twee om hemofiliepatiënt te zijn.
- Dochters van vrouwen die draagster van hemofilie zijn, hebben één kans op twee om zelf draagster te zijn.
- Dochters van een man met hemofilie zijn steeds draagster. Zij kregen namelijk één van hun twee X-chromosomen van hun vader.
- Zonen van mannen met hemofilie zijn vrij van de aandoening. Zij krijgen enkel het Y-chromosoom van hun vader, en niet het X-chromosoom met het gen-defect.
- Zonen van vrouwen die draagster zijn van hemofilie hebben één kans op twee om hemofiliepatiënt te zijn.
Naast deze familiale vorm kan hemofilie in ongeveer 30 procent van de gevallen voorkomen door een nieuwe toevallige genetische mutatie in het FVIII gen, die er dan verder voor zorgt dat hemofilie overerfbaar is binnen de familie.
Symptomen
Personen met hemofilie A bloeden niet sneller, maar wel langer. De ernst is afhankelijk van het FVIII gehalte in het bloed van de patiënt.
| graad | factorgehalte | kliniek |
|
Ernstige hemofilie |
< 1 procent |
|
|
Matig ernstige hemofilie |
? 1 procent - ?5 procent |
|
|
Milde hemofilie |
> 5 - ?50 procent |
|
Behandeling
De behandeling van hemofilie is afhankelijk van de ernst van de aandoening.
| graad | factor gehalte | behandeling |
|
Ernstige hemofilie |
< 1% |
Intraveneuze transfusie met factor VIII
|
|
Matig ernstig hemofilie |
>1-<5% |
Intraveneuze transfusie met factor VIII
|
|
Milde hemofilie |
5-40% |
Bescherming met factor VIII bij operaties |
Meestal wordt op jonge leeftijd een preventieve behandeling gestart zodra er bloedingen optreden. Hierdoor worden bloedingen voorkomen en wordt blijvende gewrichtsschade vermeden.
Er kunnen nog steeds spontane of traumatische bloedingen voorkomen. In dat geval dient de patiënt een of meerdere bijkomende behandelingen(en) met factor VIII toe. Omdat de afbraak van factor VIII in het bloed erg snel verloopt, moeten deze behandelingen met stollingsfactor vaak meerdere keren per week worden uitgevoerd.
Hemofilieremmers (inhibitoren)
Soms maakt het lichaam 'remmers' aan, die we ook wel inhibitoren of antistoffen noemen. Deze antistoffen breken factor VIII af, waardoor de bloedstolling in het gedrang komt.
Het risico op de vorming van deze remmers is groter bij hemofilie A dan bij hemofilie B.
Verschillende factoren kunnen een rol spelen bij de vorming van deze remmers. Er zijn een aantal patiëntgebonden factoren zoals:
- ernst van de hemofilie
- genetische mutatie
- etnische afkomst
- familiale voorgeschiedenis
- …
Als deze remmers optreden is een intensieve en goede opvolging door het referentiecentrum noodzakelijk.
Aandachtspunten
- ‘Factor First’ is het eerste aandachtspunt bij bloedingen van een hemofiliepatiënt. Hoe sneller een behandeling wordt opgestart, hoe kleiner de kans op grote en laattijdige letsels
- Rest, Ice, Compression, Elevation (kortom RICE) zijn belangrijke elementen in een aanvullende behandeling:
- RUST
- IJS
- COMPRESSIE
- ELEVATION/HOOGSTAND
- Hemofiliepatiënten mogen geen intramusculaire injecties toegediend krijgen. Ook vaccinaties worden best subcutaan toegediend. Indien er twijfel bestaat over de toedieningswijze van een geneesmiddel, neemt u best contact op met uw hemofiliebehandelaar.
- Koortswerende of pijnmedicatie die de bloedstolling kunnen beïnvloeden moet vermeden worden. Hieronder vallen onder andere:
- aspirinehoudende medicatie
- NSAID’s (niet steroïdale anti-inflammatoire drugs).
Bespreek tijdens de consultatie met uw hemofiliebehandelaar welke medicatie er wel kan genomen worden in het kader van verhoogde lichaamstemperatuur en pijnbehandeling. Medicatie op basis van paracetamol krijgt meestal de voorkeur.
Logboek
In geval van behandeling met FVIII-preparaten is het belangrijk dat lotnummers, hoeveelheden, reden en datums worden geregistreerd. De mogelijkheden die hiervoor worden aangeboden door het hemofiliecentrum van UZ Leuven zijn :
- Logboekje van de AHVH – registratie op papier, te verkrijgen bij de dienst.
- Mynexuz: onze beveiligde webtoepassing waar u uw afspraken, facturen en persoonlijke gegevens kunt raadplegen en een logboek kunt invullen. De gegevens van dit logboek worden direct ingeladen in uw medisch dossier.
Voor uw hemofiliebehandelaar is het van groot belang, omwille van veiligheidsoverweging maar ook omwille van therapeutische redenen, zicht te hebben op uw behandeling. Breng daarom uw logboek steeds mee wanneer u op raadpleging komt.

